Overview
The goal of the focus area is to develop theory and computational methods for complex data and systems in medicine and healthcare. We focus on scaling up computation for large numbers of genomes, machine learning methods for medical big data, as well as complex network modelling and mining. We collaborate with top medical research groups and hospitals to bring our tools towards practical use in healthcare decision support systems.
Research Groups
Algorithmic Bioinformatics (Focus Area Leader:
Veli Mäkinen, University of Helsinki; Juha Kärkkäinen, Simon Puglisi, Leena Salmela, Alexandru Tomescu)
Machine Learning for Health (Aalto-ML4H) (Focus Area Deputy Leader, group leader: Pekka Marttinen, Aalto University)
Kernel Methods, Pattern Analysis and Computational Metabolomics (KEPACO) (HIIT Deputy Director, group leader: Juho Rousu, Aalto University)
Bioinformatics and Evolution (group leader: Ville Mustonen, University of Helsinki)
Complex Systems (group leader: Jari Saramäki, Aalto University)
Research Highlights
Algorithmic Foundations of Pangenomics
String algorithms have been in vital role in the genome analysis tasks enabled by high-throughput sequencing data and the human reference genome. Recent trend in genomics is to use variation graphs to represent the genome content of a sample of individuals from the population (so-called pangenome). In our work, we have studied algorithmic foundations of pangenomics, extending string algorithms to work on graphs and related representations. Our seminal work on extending the Burrows-Wheeler transform to graphs (Sirén, Välimäki, Mäkinen, 2014) has created a new theory branch of Wheeler graphs, and it has influenced the design of popular software tools (vg, hisat2) published in Nature Biotechnology. More recently, we have e.g. tailored sparse dynamic programming algorithms to variation graphs (Mäkinen et al., 2019), shown that exact string matching is as hard as approximate string matching on graphs (Equi et al. 2019), and developed linear time segmentation algorithms to construct compressed pangenome representations in the form of founder sequences and indexable founder block graphs (Equi et al. 2023).

J. Sirén, N. Välimäki and V. Mäkinen. Indexing Graphs for Path Queries with Applications in Genome Research, in IEEE/ACM TCBB, 11(2):375-388, 2014. Earlier in WABI 2011.
V. Mäkinen, I. Tomescu, A. Kuosmanen, T. Paavilainen, T. Gagie, R. Chikhi. Sparse Dynamic Programming on DAGs with Small Width. ACM Trans. Algorithms 15(2): 29:1-29:21, 2019. Earlier in RECOMB 2018.
M. Equi, V. Mäkinen, A. I. Tomescu, R. Grossi. On the Complexity of String Matching for Graphs. ACM Trans. Algorithms 19(3): 21:1-21:25, 2023, Earlier in Proc. ICALP 2019: 55:1-55:15.
M. Equi, T. Norri, J. Alanko, B. Cazaux, A. I. Tomescu, V. Mäkinen. Algorithms and Complexity on Indexing Founder Graphs. Algorithmica 85(6): 1586-1623, 2023.
Causal modeling of blood glucose dynamics
Understanding how blood glucose is modulated by meals is central for managing blood glucose on health levels, and estimating these effects from observational time-series data from wearable devices poses a major data analysis challenge. In these two papers we developed probabilistic models, combining Gaussian processes, point processes, and causal inference to estimate interventional and counterfactual trajectories after changes in dietary habits. Furthermore, we estimated the direct and indirect effects of a gastric bypass surgery on blood glucose, where the indirect effect is mediated through changes in diet.
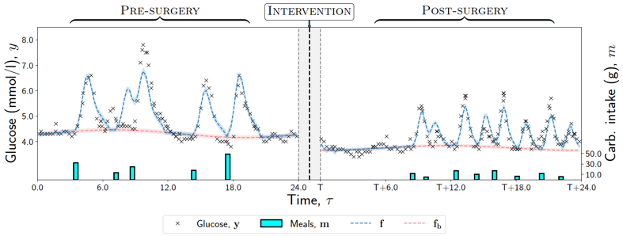
Hizli, C., et al. (2023). Temporal causal mediation through a point process: direct and indirect effects of healthcare interventions. Thirty-seventh Conference on Neural Information Processing Systems (NeurIPS 2023).
Hizli, C., et al. (2023). Causal Modeling of Policy Interventions From Treatment–Outcome Sequences. In Proceedings of the 40th International Conference on Machine Learning. (ICML 2023).
Machine learning for small molecule identification
We have developed award-winning (CASMI competitions 2016 & 2017, http://casmi-contest.org/2017/index.shtml) methods and tools for identifying and annotating small molecules from their LC-MS/MS spectra using machine learning. Our tools, developed in collaboration with Böcker group at University of Jena (CSI:FingerID, Dührkop et al. 2015; SIRIUS, Dührkop et al. 2019; CANOPUS, Dührkop et al. 2021) are widely used in the metabolomics community, e.g. our CSI:FingerID server has answered more than 120M metabolite identification queries to date. Recent improvements include a novel approach to jointly identify a set of metabolites in a biological sample giving a state of the art accuracy (Bach et al. 2022).

Dührkop et al. 2015. Searching molecular structure databases with tandem mass spectra using CSI: FingerID. Proceedings of the National Academy of Sciences, 112(41), pp.12580-12585.
Dührkop et al. 2019. SIRIUS 4: a rapid tool for turning tandem mass spectra into metabolite structure information. Nature methods, 16(4), pp.299-302.
Dührkop, et al., 2021. Systematic classification of unknown metabolites using high-resolution fragmentation mass spectra. Nature Biotechnology, 39(4), pp.462-471.
Bach, E., Schymanski, E.L. and Rousu, J., 2022. Joint structural annotation of small molecules using liquid chromatography retention order and tandem mass spectrometry data. Nature Machine Intelligence, 4(12), pp.1224-1237.
Fast and Minimum Representations of K-mer Sets
De Bruijn graph is a fundamental data structure in genomics, representing relationships of length k substrings. We have developed the fastest de Bruijn graph builder to date (Cracco and Tomescu, 2023), as well as found a linear-time algorithm to construct the minimum plain text representation for a set of length k substrings (Schmidt and Alanko, 2023). This latter problem was earlier conjectured to be NP-hard. We also developed a polynomial-time solution to the multiset variant (Schmidt et al., 2023).
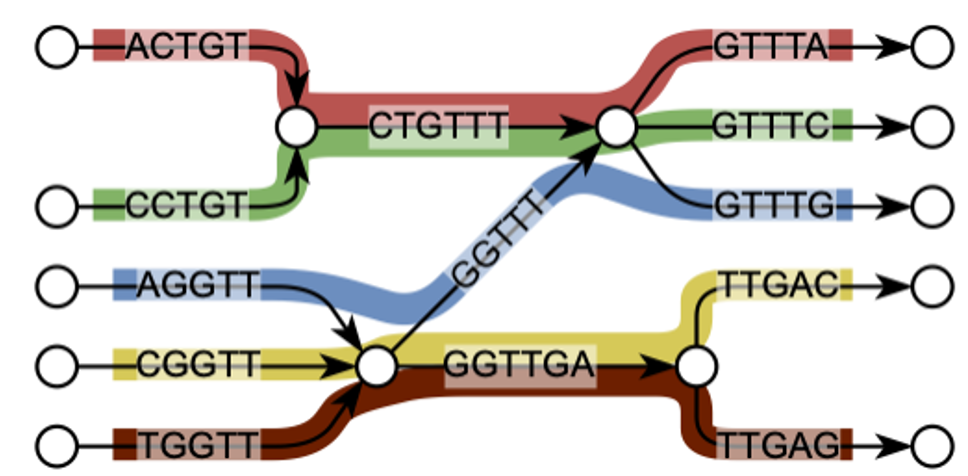
Andrea Cracco and Alexandru I. Tomescu. Extremely fast construction and querying of compacted and colored de Bruijn graphs with GGCAT. Genome Research, 2023.
Sebastian Schmidt and Jarno N. Alanko. Eulertigs: minimum plain text representation of k-mer sets without repetitions in linear time. Algorithms for Molecular Biology, 18:5 (2023). Best paper award at WABI 2022.
Sebastian Schmidt, Shahbaz Khan, Jarno N. Alanko, Giulio E. Pibiri, and Alexandru I. Tomescu. Matchtigs: minimum plain text representation of k-mer sets. Genome Biology, 24:136, 2023.
Improved pipeline for constructing functional brain networks from fMRI data
In complex network modelling and mining applied to neuroscience, we have made a significant push towards better and more informed modelling and interpretation of functional brain networks derived from fMRI time series. In a series of papers (Korhonen et al. 2017, Alakörkkö et al. 2017, Ryyppö et al. 2018, Triana et al. 2019) we have focused on the preprocessing and modelling pipeline and shown that i) commonly applied spatial smoothing has unexpected effects and may give rise to artefacts, ii) the common approach of defining RoIs for functional network construction also suffers from problems; RoIs may require a dynamic interpretation.
O Korhonen et al 2017. Consistency of regions of interest as nodes of fMRI functional brain networks. Network Neuroscience 1 (3), 254-274 (2017)
Alakörkkö et al. 2017. Effects of spatial smoothing on functional brain networks. European Journal of Neuroscience 46 (9), 2471-2480 (2017)
Ryyppö et al. 2018. Regions of Interest as nodes of dynamic functional brain networks. Network Neuroscience 2 (4), 513-535 (2018)
Triana et al. 2019. Effects of spatial smoothing on group-level differences in functional brain networks. Network Neuroscience 4 (3), 556-574 (2019)
Predicting drug combination responses in cancer and infectious diseases
We have developed new machine learning methods for high-throughput screening of drug combinations for cancer therapies, including cost-effective experimental design of pairwise combination matrices (DECREASE, Ianevski et. al, 2019) and accurate prediction of dose-response matrices in cancer cell lines (comboFM, Julkunen et al. 2020; comboLTR, Wang et al. 2021). Current research includes prediction of higher-order drug interaction effects using higher-order tensors, as well translating the pre-clinical model predictions to cancer patients using transfer learning.

Ianevski, et al. 2019. Prediction of drug combination effects with a minimal set of experiments. Nature machine intelligence, 1(12), pp.568-577.
Julkunen et al, 2020. Leveraging multi-way interactions for systematic prediction of pre-clinical drug combination effects. Nature communications, 11(1), pp.1-11.
Wang et al. 2021. Modeling drug combination effects via latent tensor reconstruction, Bioinformatics, Volume 37, Issue Supplement_1, July 2021, Pages i93–i101,
Eco-evolutionary control of pathogens
Predictability and control of evolving populations is an emerging topic of high scientific interest and vast translational potential in applications such as vaccine and therapy design. Here we have started to developed eco-evolutionary control theory. Successful control of eco-evolutionary trajectories hinges on the intrinsic dynamics of the target system and the control leverage that can be applied. For example, we have studied what are the key determinants of controllability and how to apply control to improve therapies against pathogens.
M. Lässig and V. Mustonen, 2020. Eco-evolutionary control of pathogens. Proceedings of the National Academy of Sciences, 117(33), pp.19694-19704.